mRNA B0511.1 Chr1:1..100 Type=UTR;Note="Putative primase" mRNA B0511.1 Chr1:101..200,300..400,500..800 Type=CDS mRNA B0511.1 Chr1:801..1000 Type=UTRThe top level feature's primary tag will be "mRNA", as indicated in the first column. It will contain five subparts, a 5' UTR spanning positions 1..100, a series of three CDS (coding) regions, and a 3' UTR extending from positions 801..1000.
Additional tags that are placed in the first line of the feature, such as the Note, will be applied to the top level. In this example, the note "Putative primase" will be applied to the mRNA at the top level of the feature:
Grouping
You can group related features together. The layout will attempt to keep grouped features together, and will connect them with a dotted or solid line if the connector option is specified.
A group is created using a line that contains just two columns consisting of the feature type and name. This is followed by a series of data lines in which the feature type is blank. For example:
cDNA-clone yk53c10
yk53c10.5 18892-19154
yk53c10.3 15000-15500,15700-15800
[cDNA-clone]
glyph = segments
connector = dashed
This example creates a group of type "cDNA-clone" named Yk53c10. It consists of two sub-features, one the 5' EST and the other the 3' EST. The two configuration section that follows this group says to use the "segments" glyph and to connect the parts using a dashed line. This is described in more detail later.
You can add URLs and descriptions to the components of a group, but not to the group as a whole.
Comments
You can place a comment in the annotation file by preceding it with a pound sign (#). Everything following the pound sign is ignored:
# this is a comment
Customizing the Display
The browser will generate a reasonable display of your annotations by default. However, when using either FFF or GFF3 formats, you can customize the appearance extensively by including one or more configuration sections in the annotation file. In addition to changing the size, color and shape of the graphical elements, you can attach URLs to them so that the user will be taken to a web page of your choice when he clicks on the feature.
Here is an example configuration section. It can appear at the top of xthe file, at the bottom, or interspersed among data sections:
# example file [general] height = 12 strand_arrow = 1 [EST] glyph = segments bgcolor= yellow connector = dashed height = 5 [FGENES] glyph = gene bgcolor = green description = 1
The configuration section is divided into a set of sections, each one labeled with a [section title]. The [general] section specifies global options for the entire image. Other sections apply to particular feature types. In the example, the configuration in the [EST] section applies to features labeled as having type "EST", while the configuration in the [FGENES] section applies to features labeled as predictions from the FGENES gene prediction program. Options in more specific sections override those in the general section.
Inside each section is a series of name=value pairs, where the name is the name of an option to set. You can put whitespace around the = sign to make it more readable, or even use a colon (:) instead if you prefer. The following option names are recognized:
| Option | Value | Example |
|---|---|---|
| bgcolor | Background color of each element | blue |
| bump | Prevent features from colliding (0=no, 1=yes) | 1 |
| connector | Type of group connector (dashed, hat or solid) | dashed |
| description | Whether to print the feature's description (0=no, 1=yes) | 0 |
| fgcolor | Foreground color of each element | yellow |
| glyph | Style of each graphical element (see list below) | transcript |
| height | Height of each graphical element (pixels) | 10 |
| key | Key to the feature. This is a human-readable description that will be printed in the key section of the display | ESTs aligned via TwinScan 1.2 |
| label | Print the feature's name (0=no, 1=yes) | 1 |
| linewidth | Width of lines (pixels) | 3 |
| link | URL to link to. This is a Web link in which certain variables beginning with the "$" will be replaced with feature attributes. Recognized variables are: $name - the name of the feature, and $type - the type of the feature (e.g. EST). | link = http://www.your.site/db/get?id=$name;type=$type |
| citation | This is a longer narrative description of the feature intended to identify the author and detailed description of the method. It can be either a text description or a link. | http://your.site.org/detailed_description.html |
| strand_arrow | Indicate feature strandedness using an arrow (0=no, 1=yes). NB: Strandedness is depicted differently by different glyphs, and in some cases is the default. | 1 |
| section | Indicates where in the gbrowse window this type of feature
should be placed: "details"=details panel;
"overview"=overview panel; "region"=region panel (if there is
one for this source);
"details+overview"=both panels; "details+overview+region"=all
three panels. "details" is the default. |
details+overview |
The bump option is the most important option for controlling the look of the image. If set to false (the number 0), then the features are allowed to overlap. If set to true (the number 1), then the features will move vertically to avoid colliding. If not specified, bump is turned on if the number of any given type of sequence feature is greater than 50.
Unlike the data section, you do not need to put quotes around option values that contain white space. In fact, you can continue long option values across multiple lines by putting extra space in front of the continuation lines:
[GenomeAlign] citation = The pseudoobscura genome was aligned to melanogaster using GenomeAlign version 1.0. High-similarity regions are shown in blue, low similarity regions are shown in orange. The work was performed by Joe Postdoc, and is currently in press.
Some glyphs also have glyph-specific options. These are described in detail below.
Colors
Colors are English-language color names or Web-style #RRGGBB colors (see any book on HTML for an explanation). The following colors are recognized:
| white | coral | darkslateblue | green | lightpink | mediumslateblue | paleturquoise | sienna |
| black | cornflowerblue | darkslategray | greenyellow | lightsalmon | mediumspringgreen | palevioletred | silver |
| aliceblue | cornsilk | darkturquoise | honeydew | lightseagreen | mediumturquoise | papayawhip | skyblue |
| antiquewhite | crimson | darkviolet | hotpink | lightskyblue | mediumvioletred | peachpuff | slateblue |
| aqua | cyan | deeppink | indianred | lightslategray | midnightblue | peru | slategray |
| aquamarine | darkblue | deepskyblue | indigo | lightsteelblue | mintcream | pink | snow |
| azure | darkcyan | dimgray | ivory | lightyellow | mistyrose | plum | springgreen |
| beige | darkgoldenrod | dodgerblue | khaki | lime | moccasin | powderblue | steelblue |
| bisque | darkgray | firebrick | lavender | limegreen | navajowhite | purple | tan |
| blanchedalmond | darkgreen | floralwhite | lavenderblush | linen | navy | red | teal |
| blue | darkkhaki | forestgreen | lawngreen | magenta | oldlace | rosybrown | thistle |
| blueviolet | darkmagenta | fuchsia | lemonchiffon | maroon | olive | royalblue | tomato |
| brown | darkolivegreen | gainsboro | lightblue | mediumaquamarine | olivedrab | saddlebrown | turquoise |
| burlywood | darkorange | ghostwhite | lightcoral | mediumblue | orange | salmon | violet |
| cadetblue | darkorchid | gold | lightcyan | mediumorchid | orangered | sandybrown | wheat |
| chartreuse | darkred | goldenrod | lightgoldenrodyellow | mediumpurple | orchid | seagreen | whitesmoke |
| chocolate | darksalmon | gray | lightgreen | mediumseagreen | palegoldenrod | seashell | yellow |
| coral | darkseagreen | green | lightgrey | mediumslateblue | palegreen | sienna | yellowgreen |
Glyphs
The ``glyph'' option controls how the features are rendered. The following glyphs are implemented:
| Name | Description | |
|---|---|---|
| generic | A filled rectangle. | |
| ellipse | An oval | |
| arrow | An arrow; can be unidirectional or bidirectional. It is also capable of displaying a scale with major and minor tickmarks, and can be oriented horizontally or vertically. | |
| segments | A set of filled rectangles connected by solid lines. Used for interrupted features, such as gapped alignments and exon groups. | |
| gene | The "gene" glyph is suitable for drawing coding genes. The coding regions will be drawn using the specified bgcolor, and the UTRs will be drawn in grey. You can change the color of the UTRs by specifying a "utr_color" option. For the gene glyph to work properly, the top level feature must be of type "mRNA" and the subparts of type "UTR", "five_prime_UTR", "three_prime_UTR", or "CDS". See the top of this document for an example. | |
| transcript | Similar to segments, but the connecting line is a "hat" shape, and the direction of transcription is indicated by a small arrow. | |
| transcript2 | Similar to transcript, but the direction of transcription is indicated by a terminal segment in the shape of an arrow. | |
| anchored_arrow | Similar to arrow, but the arrow is drawn in order to take account of features whose end-point(s) are unknown, rather than to indicate strandedness. | |
| primers | Two inward pointing arrows connected by a line. Used for STSs. | |
| triangle | A triangle, used to represent point features like SNPs, or deletions and insertions. May be oriented north, south, east or west. | |
| xyplot | A histogram, line plot or column chart,
used for graphic numeric features such as microarray intensity values. To indicate
the value you wish to chart, add a score=XXXX note to the description section:
| |
| wiggle_xyplot | Similar to the xyplot glyph, but specialized for displaying very dense quantitative data. When you upload a WIG file, this glyph is automatically chosen for you. | |
| graded_segments | A set of connected segments whose colors change intensity according to a score indicated by a "score=XXX" tag. The low and high scores are indicated by "min_score" and "max_score" options in the configuration stanza, and the basic color is indicated by "bgcolor." | |
| wiggle_density | Similar to the graded_segments glyph, but specialized for displaying very dense quantitative data. When you upload a WIG file, this glyph is automatically chosen for you as an alternative to wiggle_xyplot. | |
| heat_map | A set of connected segments whose colors change hue according to a score indicated by a "score=XXX" tag. The low and high scores are indicated by "min_score" and "max_score" options in the configuration stanza, and the start and ending hues are indicated by "start_color" and "end_color." A feature with score equal to min_score will be displayed using start_color, while a feature with score equal to max_score will be displayed using end_color. Intermediate scores are displayed by blending the two hues. | |
| trace | Reads a SCF sequence file and displays the trace graph. For this glyph to
work, the trace file must be placed on a web-accessible FTP or HTTP server and the
location indicated by a "trace" tag:
|
The following glyph-specific options are recognized:
| Glyph | Option | Description |
|---|---|---|
| arrow, anchored_arrow | tick | Draw major and minor ticks on arrow. (0 = no ticks, 1 = major ticks, 2 = major & minor ticks) |
| parallel | Whether to draw the arrow parallel to the sequence or perpendicular to it (1=parallel, 0=antiparallel). | |
| northeast, east | Force a north or east arrowhead. (The two option names are synonyms.) (0=false, 1=true) | |
| southwest, west | Force a south or west arrowhead. (The two option names are synonyms.) (0=false, 1=true) | |
| double | force doubleheaded arrow (0=false, 1=true) | |
| base | Draw a vertical base at the non-arrowhead side (0=false, 1=true). | |
| scale | Reset the labels on the arrow to reflect an externally established scale. | |
| gene | utr_color | Color of the UTRs |
| thin_utr | If set to a non-zero value, then UTRs will be drawn as thin boxes | |
| decorate_introns | If set to a non-zero value, then introns will be decorated with little arrows indicating the direction of the transcript | |
| primers | connect | Whether to connect the inward-pointing arrowheads by a line (0=false, 1=true) |
| connect_color | Color of the connecting line | |
| triangle | point | Is this a point-like feature? If true, the triantle will be drawn at the center of the range, and not scaled to the feature width. (0=false, 1=true) |
| orient | Orientation of the triangle. (N=north, S=south, E=east, W=west) | |
| xyplot, wiggle_xyplot | graph_type | Type of graph (histogram, boxes, line, points, linepoints) |
| min_score | Minimum score for feature (will be level 0 on graph) | |
| max_score | Maximum score for feature (will be level 0 on graph) | |
| scale | Where to draw the Y axis scale, if any (left, right, both, none) | |
| point_symbol | When using points or linepoints graph types, controls the symbol to use for the data points. One of triangle, square, disc, point, or none. | |
| point_radius | The radius of the symbols, if applicable, in pixels | |
| bicolor_pivot | This is a numeric option, which, if specified, causes the histogram to be drawn in two colors, one for values above the limit specified by the "pos_color" option and one for values below the limit specified by the "neg_color" option. | |
| pos_color | The color to draw values which are above bicolor_pivot, if bicolor_pivot is specified. | |
| neg_color | The color to draw values which are below bicolor_pivot, if bicolor_pivot is specified. | |
| graded_segments, wiggle_density | min_score | Minimum score for the feature (will be drawn as a white segment) |
| max_score | Maximum score for the feature (will be drawn as a segment with full intensity bgcolor) | |
| heat_map | min_score | Minimum score for the feature (the segment will be drawn with the starting hue) |
| max_score | Maximum score for the feature (the segment will be drawn with the ending hue) | |
| start_color | Color for segments with the minimum score | |
| end_color | Color for segments with the maximum score | |
| bicolor_pivot | This is a numeric option, which, if specified, causes the histogram to be drawn in two colors, one for values above the limit specified by the "pos_color" option and one for values below the limit specified by the "neg_color" option. | |
| pos_color | The color to draw values which are above bicolor_pivot, if bicolor_pivot is specified. | |
| neg_color | The color to draw values which are below bicolor_pivot, if bicolor_pivot is specified. | |
| trace | trace | Specify the trace path or URL to use for this feature. |
| trace_prefix | String to prepend to each trace path. You may prepend a directory or a partial URL. | |
| trace_height | The height in pixels that the trace will be drawn. | |
| vertical_spacing | Vertical distance from the box that shows the physical span of the feature to the top of the picture (in pixels). | |
| glyph_delegate | Glyph to use when zoomed out too far for the trace to be drawn. | |
| a_color | Color of the line representing Adenine on the trace. | |
| c_color | Color of the line representing Cytosine on the trace. | |
| g_color | Color of the line representing Guanine on the trace. | |
| t_color | Color of the line representing Thymine on the trace. | |
| show_border | Show the black border from around the trace (0=false, 1=true) |
Displaying Intensity Plots & Other Numeric Data
For large data sets such as chromosome-wide tiling arrays, please use Wiggle (WIG) format. However, for smaller data sets (1-10,000 points), you can use FFF format to achieve a quick and dirty display.
Here is a simple template for you to follow. The result is shown on
the right.
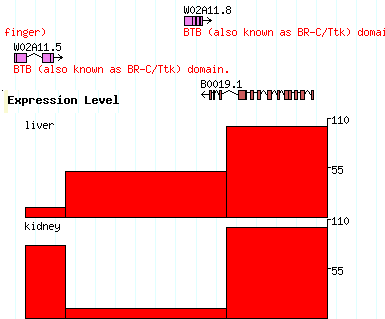
[expression] glyph = xyplot graph_type=boxes fgcolor = black bgcolor = red height=100 min_score=0 max_score=110 label=1 key=Expression Level reference=chr1 expression liver 10000..12000 score=10 expression liver 12001..19999 score=50 expression liver 20000..25000 score=100 expression kidney 10000..12000 score=80 expression kidney 12001..19999 score=10 expression kidney 20000..25000 score=100 |
The [expression] section says to use the xyplot type of glyph, to set its type to "boxes" (a column chart), to make the fore and background colors black and red respectively, to set the height of the chart to 100, and to set the minimum and maximum values for the Y axis to 0 and 110 respectively. We also add a label and a human-readable track key.
The data section defines two experiments to show in the track. Both experiments use probes whose positions are relative to landmark B0019 (you can of course use chromosome coordinates, or whatever you choose). Both experiments are of type "expression", but one is the "liver" experiment and the other is the "kidney" experiment, as indicated in the second column. The third column contains the coordinates of each assay point, and the fourth column contains the score=XXX attribute, where XXX is the intensity value.
See the chart in the previous section for the xyplot glyph options.
Binary data file formats
Binary data file formats provide a convenient, efficient, and rapid access to genomic data, either segments or quantitative data. They are indexed, allowing for immediate random access to any location, and compressed for disk space savings. The following file types are accepted:
- Bam file
- The Bam file is a binary version of the text SAM format, which is a sequence alignment file for next generation sequencing technologies. Bam files usually contain millions of alignments of short sequence reads. They may be displayed in one of two ways: as a collection of segments with or without the DNA sequence of the read (depending on zoom level), or as a quantitative xyplot representing the coverage or depth of sequencing. Bam files may be generated, sorted, and/or indexed from Sam files using samtools or Picard. Bam files have a ".bam" extension. Displaying Bam files requires the installation of the Bio::DB::Sam Perl module.
- bigWig file
- The bigWig file is a binary version of the text Wiggle format for displaying dense quantitative data. BigWig files are generated from text wiggle files, either fixedStep, variableStep, or bedGraph variants, using either the wigToBigWig or bedGraphToBigWig utilities available from UCSC. File conversion requires a file with the name and lengths of the chromosomes for your genome version. Such a file may be obtained by selecting the Download Chrom Sizes from the "File" menu in the upper left corner of the genome browser page. BigWig files usually have a ".bw" extension. Displaying bigWig files requires the installation of the Bio::DB::BigWig Perl module.
- Archive of bigWig files
- Two or more bigWig files may be combined as a BigWigSet collection, providing a fast, convenient method of transferring a related collection of genomic data files and grouping them into a single track, with each bigWig file represented as a subtrack selectable by display name. Supported archive formats include TAR and ZIP files. Two or more bigWig files (with a .bw extension) and optionally a metadata text file may be included. Extraneous files and directory paths are ignored.
- bigBed file
- The bigBed file is a binary version of the text BED format for displaying dense genomic regions or intervals. Data from bigBed files may be displayed as either segments or as a quantitative xyplot representing coverage (the depth of segments at any given locus). BigBed files are generated from text BED files using the bedToBigBed utility available from UCSC. File conversion requires a file with the name and lengths of the chromosomes for your genome version. Such a file may be obtained by selecting the Download Chrom Sizes from the "File" menu in the upper left corner of the genome browser page. BigBed files usually have a ".bb" extension. Displaying bigBed files requires the installation of the Bio::DB::BigBed Perl module.
- useq file
- The useq file is a compressed archive of either genomic intervals (with optional scores and/or text) or quantitative data. It is generated using utilities from the USeq analysis package. Upon upload, the useq file is automatically converted to either a bigWig file or bigBed file depending on context. Processing useq files requires the installation of the USeq package, wigToBigWig and bedToBigBed utilities, and the Bio::DB::BigWig Perl module.
To import binary files, generate the files using the appropriate utility and ensure they have the appropriate file extension. Under the "Custom Tracks" tab, select the "From a file" link, click the "Choose file" button, and navigate to your file using the Dialog box. Click "Upload" to upload the file. Status reports may be printed as the file is uploaded and processed. Successfully uploaded files will be displayed under the "Custom Tracks" and "Select Tracks" tabs.
A basic configuration will be generated appropriate for the file type uploaded. The configuration may be edited by selecting the "edit" link adjacent to the configuration file.
Importing Custom Tracks via the Internet
GBrowse can display annotation files that are physically located on internet-connected sites. Use the "Import a track" section to paste in the URL of an annotation file. The following URL types are allowed:
- The URL of a BED, GFF3, or FFF file.
- If a BED, GFF3 or FFF file is located on an internet-accessible FTP or web server, paste its URL into the "remote URL" text field. It will be mirrored to the GBrowse server and displayed. When you update the file, the updated version will be mirrored automatically.
- The URL of a BigWig (.bw) file
- The URL of a remote bigWig file may be provided. The file must have a ".bw" extension and be located on an FTP or Web server that can be accessed via this browser. Paste the full URL of the BigWig file into the "Import a track URL" field and press "upload".
- The URL of a BigBed (.bb) file
- The URL of a remote bigBed file may be provided. The file must have a ".bb" extension and be located on an FTP or Web server that can be accessed via this browser. Paste the full URL of the bigBed file into the "Import a track URL" field and press "upload".
- The URL of a BAM file
- A sorted, indexed Bam file may be accessed remotely. Both the Bam file with a ".bam" extension and its corresponding ".bai" index file must located on Web or FTP server. Paste the full URL of the Bam file (NOT the .bai file) into the address field "From a URL" link Place the BAM file and its associated .bai index file on a web or FTP-accessible server and paste its URL into the "Import a track URL" field. The information in the file will be retrieved as needed in a network-efficient manner. It is up to you to ensure that the chromosome coordinates of the BAM file match the build of the genome used by this instance of the browser, or nothing will show up (you'll get blank tracks). Please be aware that the URL must end with ".bam" for GBrowse to recognize that it is a BAM file.
- A GBrowse or DAS URL
- You can share tracks from one GBrowse
server to another by clicking on the
 icon. This will give you a URL
that you can paste into the URL import field. Alternatively, you
can view a large variety of annotations using the distributed
annotation system (DAS) protocol. The DAS registry will help you
locate DAS servers with useful genomic data. Then cut and paste the
DAS server URL into the remote track URL box as before.
icon. This will give you a URL
that you can paste into the URL import field. Alternatively, you
can view a large variety of annotations using the distributed
annotation system (DAS) protocol. The DAS registry will help you
locate DAS servers with useful genomic data. Then cut and paste the
DAS server URL into the remote track URL box as before.
$Id$